提到“视黄醛(A醛)”,首先会想到什么?当然是抗老!!!A酸、A醇、A醛、玻色因......,但是,“视黄醛”另一个作用却鲜为人知,视黄醛缺乏——糖尿病恶化。
视黄醛(Rald,A醛)是视黄醇转化为视黄酸(RA,A酸)过程的中间物,属于维生素A类物质,是机体维持正常生理过程的必需营养素。近年来,越来越多研究发现Rald可以影响机体糖脂代谢平衡,此过程主要由葡萄糖激酶(GCK)、磷酸烯醇丙酮酸羧激酶1(PCK1)和葡萄糖-6-磷酸酶(G6PC)等酶控制。中国药科大学刘晓东团队前期研究中发现Rald浓度稳定对维持机体糖稳态具有重要作用,且2型糖尿病(T2D)患者体内常常伴有Rald水平降低现象,但Rald缺乏在T2D中作用的具体机制却尚不清晰。
2023年5月6日,中国药科大学刘李/刘晓东和南京第一人民医院邹建军为共同通讯作者在《Acta Pharmaceutica Sinica B》(IF=14.5)发表了题为:“Hepatic retinaldehyde deficiency is involved in diabetes deterioration by enhancing PCK1- and G6PC-mediated gluconeogenesis”的研究论文,在这项研究中,作者通过添加Rald/RA或利用AAV9-RALDH1-shRNA使肝脏中视黄醛脱氢酶1(RALDH1)表达下降,显著降低了T2D小鼠的空腹血糖水平,并上调了Rald表达,下调了PCK1和G6PC的表达,证明了肝脏Rald缺乏与T2D恶化相关,并且在高脂饮食喂养的小鼠、人原代肝细胞、油酸(OA)处理的人肝癌细胞(HepG2)中也证明了这一点。利用荧光素酶报告实验和分子对接,从机制上解释了肝脏Rald缺乏、肝脏糖异生和T2D恶化之间的联系,同时也证明了Rald是T2D潜在治疗靶点,为T2D治疗提供了新的理论支撑。
划重点!!!本研究使用了汉恒生物提供的针对RALDH1基因沉默的腺相关病毒(AAV9-RALDH1-shRNA)。

下面,介绍一下具体的研究过程:
首先,作者对T2D小鼠体重,采食量,饮水量以及空腹血糖量等生理参数进行检测发现T2D小鼠往往伴随着肝脏糖异生作用增强和葡萄糖利用受损的情况,并且肝脏Rald表达量降低RALDH1表达量升高。而在Rald或Rald+RALDH1抑制剂作用后,Rald表达量显著升高,小鼠的糖尿病症状得到缓解,表明小鼠肝脏Rald缺乏与T2D恶化相关。

图1. 肝脏Rald缺乏导致T2D恶化
为了探究RALDH1表达量下降是否与T2D恶化相关,作者利用AAV9-RALDH1-shRNA对T2D小鼠肝脏RALDH1基因进行特异性沉默,结果发现RALDH1基因沉默后,小鼠肝脏Rald基因水平显著上升,小鼠的糖尿病症状得到改善,表明肝脏RALDH1沉默可以增加肝脏Rald水平从而改善肝脏糖异生。

图2. RALDH1沉默通过增加肝脏Rald水平缓解肝糖异生
据报道,T2D肝脏糖异生占肝脏葡萄糖产量的75%,此过程主要受GCK、PCK1和G6PC等基因的调控。作者检测T2D小鼠中PCK1、G6PC和GCK表达水平发现呈增加趋势,Rald+RALDH1抑制剂作用后PCK1和G6PC表达量降低,GCK表达量增加,沉默肝脏RALDH1基因后,与对照组相比T2D小鼠中PCK1和G6PC表达水平均下降,GCK蛋白水平未受影响,因此证明PCK1和G6PC表达量的改变可能是导致肝脏Rald水平变化的原因。

图3. PCK1和G6PC表达量变化影响肝脏Rald水平
为了探究正常小鼠Rald缺乏是否影响肝脏糖代谢,作者采用C57BL/6J小鼠并配合高脂饮食,检测结果表明高脂饲养的小鼠体重、空腹血糖和腹腔注射葡萄糖或丙酮酸后的血清中葡萄糖含量上升,Rald表达量下降,表明小鼠肝脏糖异生作用增强,利用AAV9-RALDH1-shRNA沉默高脂饲养小鼠RALDH1基因后,Rald表达量显著升高,相应的PCK1和G6PC表达量下降,小鼠腹腔注射葡萄糖或丙酮酸后的血清中葡萄糖含量相较高脂饲养小鼠有所下降,但是体重不变,说明RALDH1沉默减轻高脂饮食小鼠肝脏糖异生。
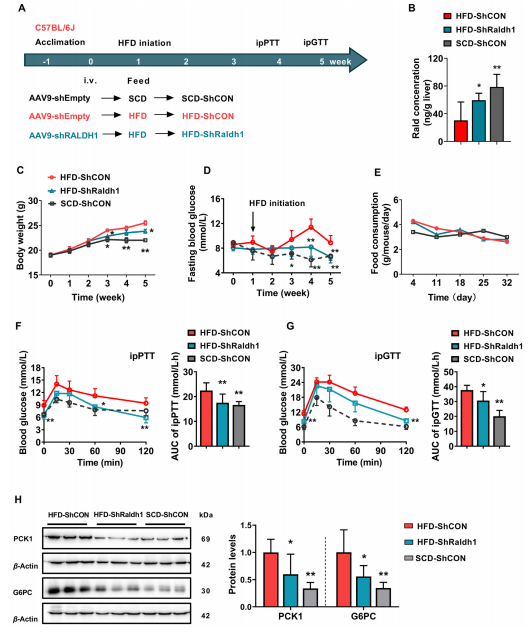
图4. RALDH1沉默减轻高脂饮食小鼠肝脏糖异生
大量研究发现,T2D通常与肝脏脂肪变性有关,因此作者利用油酸(OA)处理HepG2细胞探究Rald在肝脏糖异生中的作用。显微镜下观察OA处理的HepG2细胞出现肝脏脂肪变性的特征,细胞胞浆内出现脂滴并且伴随葡萄糖利用受损以及葡萄糖产量增加,G6PC和PCK1表达水平也显著高于对照组。利用siRNA下调OA处理组RALDH1表达,结果发现Rald和Rald+siRALDH1都可以显著抑制葡萄糖的产生并下调G6PC和PCK1表达,但是对葡萄糖的利用以及对GCK的表达几乎没有影响。之后作者在来自四个捐赠者的人原代肝细胞中重复实验,发现了与HepG2细胞中一样的现象,表明Rald通过抑制OA处理的HepG2细胞和原代人肝细胞中PCK1和G6PC的表达而改善肝脏糖异生。

图5. Rald通过抑制PCK1和G6PC的表达而改善肝脏糖异生
Rald的化学结构与RA相似,因此作者猜测Rald可能通过影响RA受体下调PCK1和G6PC的表达。作者使用视黄酸受体(RAR)激动剂LG100268和TTNPB刺激OA处理的HepG2,结果显示细胞中PCK1和G6PC表达上调,且能被维甲酸X受体(RXR)拮抗剂HX531和Ro41-5253抑制,但PPARγ(过氧化物酶体增殖物激活受体γ亚型)的激动剂和抑制剂均对PCK1和G6PC的表达无影响,说明RXR和RAR可能参与OA处理的HepG2细胞中PCK1和G6PC的转录调控。而后使用Rald刺激细胞,发现Rald能抑制RAR激动剂诱导的PCK1和G6PC上调,其中对LG100268的抑制效果更为显著,说明Rald抑制了RXR诱导的PCK1和G6PC的表达。CYP26A是RAR的已知靶基因,Rald和TTNPB均显著诱导CYP26A表达,并且Rald进一步增强了LG100268+TTNPB诱导的CYP26A上调,而Ro41-5253完全消除了Rald对CYP26A的诱导作用,综合来看,Rald是RAR的激动剂和RXR的拮抗剂。

图6. Rald通过拮抗RXR抑制PCK1和G6PC的表达
为探究Rald/RA对RXRa/RARa激活的影响,作者进行荧光素酶报告实验测定Rald/RA对RARa/RXRa激活情况,首先Rald和RA以浓度依赖的方式刺激RARα,结果显示Rald和RA分别激活RARα,但是Rald拮抗RA诱导的RARα激活,表明Rald是RARα的激动剂。紧接着作者利用RA、9C-RA(9顺式RA)、Rald和OA以浓度依赖的方式刺激RXRa,结果显示RA、9C-RA和OA激活RXRa,但Rald拮抗RA或者OA诱导的RXRa激活,对于此种现象作者进行分子对接以研究Rald对RXRa的拮抗作用,结果表明,Rald、RA、9C-RA和OA以不同亲和力紧密结合在RXRa配体结合口袋中(Rald>9C-RA>RA>OA),但是与其他组相比,Rald与丙氨酸-327间没有氢键,因此在RXRa的结构域中表现出独特的分子构型,RXRa结构域螺旋11外展抑制了RXRa激活物可结合表面的形成,这也是Rald对RXRa拮抗作用的证明。

图7. Rald拮抗RXRα并激活RARα
一般情况下,RXR会与核受体形成同源二聚体或异源二聚体,通过与基因不同的DR区(结构域)结合,在转录调控中发挥重要作用。在人原代肝细胞中,作者发现Rald影响SREBF1、CYP4F2和RARB2的表达,这些基因被报道受到不同DR的调节。SREBF1和CYP4F2受DR1调控,GCK和CRABP2受DR2调控,RARB2和CYP26A受DR5调控,而Rald对PCK1和G6PC调控模式,与DR1调控SREBF1和CYP4F2的调控模式相似。作者为了证明自己的发现,构建了三个荧光素酶报告系统(RXR:RXR-DR1,RAR:RXR-DR1和RAR:RXR-DR5)进行探究,荧光素酶报告分析进一步证实,Rald作为RXR的拮抗剂,通过抑制RXR:RXR-DR1的激活,下调PCK1和G6PC的表达,进一步证明肝脏Rald缺乏参与了T2D的恶化。
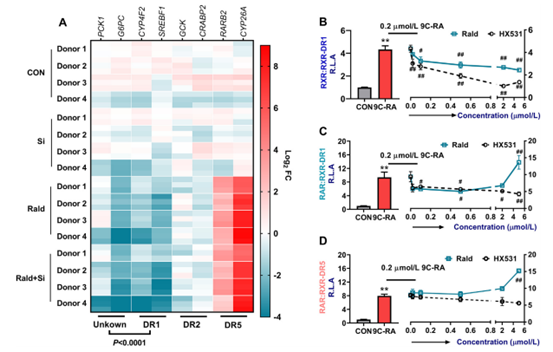
图8. Rald通过拮抗RXR:RXR-DR1的激活下调PCK1和G6PC的表达
综上所述,作者通过对T2D小鼠分析,发现T2D诱导肝脏RALDH1表达,导致肝脏Rald缺乏,Rald治疗或肝脏RALDH1沉默可以抑制T2D状态下亢进的糖异生。而Rald作为RXR的拮抗剂,在肝细胞上抑制RXR/DR1介导的PCK1和G6PC表达,同时Rald也可作为RAR的部分激动剂,维持RAR正常生理活性,对维持血糖稳态具有重要作用,肝脏Rald缺乏则增强肝脏糖异生进而加剧T2D的恶化。这些结果证明了Rald在T2D恶化中的作用,并为开发以RALDH1抑制剂或Rald为基础的调节T2D的治疗药物提供理论依据。









 3833
3833










