在上一系列的干货分享中,我们介绍了CRISPR-Cas9基因敲除系统(knock out)相关知识。接下来,我们将目光聚焦 CRISPR/cas9系统的另一应用——敲入,开启一个新的篇章。本篇文章主要从实验原理、HDR模板的选择和实验流程步骤三个方面来介绍CRISPR-Cas9基因敲入系统(knock in)。
一、CRISPR基因敲入实验原理
CRISPR-Cas9基因敲入(KI, Knock in)是指Cas9内切酶切割双链DNA时,同时提供一段与靶基因高度同源的DNA修复模板,生物体即可启动HDR修复路径,将一段外源DNA定点插入基因内部。KI的外源基因可以是具有蛋白表达功能的编码基因,可以是参与基因调控的DNA元件,也可以是一段没有功能的DNA序列等。在该修复路径中,需要将一段与预期编辑位点上下游紧邻序列具有高度同源性的DNA修复模板、特异的gRNA和Cas9核酸酶一起引入细胞中。在高度同源性DNA模板存在的情况下, HDR机制可以通过同源重组将一段DNA精准地插入特定的基因组位点。
目前CRISPR/Cas9 Gene Knock in系统可用于目的基因引入点突变,从而模拟人类遗传疾病模型;或者将报告基因(如EGFP,mCherry,BFP等)通过同源重组的方式引入目的基因的特定位点,从而可以通过报告基因的表达跟踪目标基因的表达;亦或将功能缺失的DNA片段修复为有功能的DNA片段,即可实现基因治疗的目的。此系统的应用范围不胜枚举,已成为人类生物学、医学、农业和微生物学等领域有力的基因编辑工具。
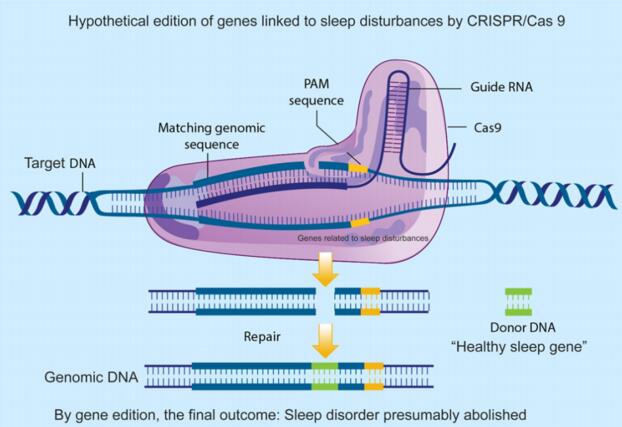
图1基于CRISPR/Cas9基因敲入的示意图(Murillo-Rodríguez E et al., 2018)
二、如何选择合适的CRISPR基因敲入HDR模板
基因敲入HDR模板的作用
在CRISPR基因敲入中,同源重组修复 (homology directed repair, HDR) 模板是一种用于指导修复DNA断裂并实现精确基因敲入的DNA序列,与目标基因组中被Cas9剪切的位置具有相同序列的同源臂。无HDR模板的CRISPR基因敲入,细胞通过非同源末端连接(non-homologous end joining,NHEJ)方式修复Cas9切割的DNA断裂。NHEJ修复方式速度快效率高,但易引起插入/缺失、突变等不良后果,故属于相对不准确的修复方式,存在错误DNA连接和变异,敲入效率和精确性较低。使用HDR模板时,细胞将利用其内在的修复机制,与模板同源臂序列进行重组,精确地修复Cas9切割的DNA断裂并实现精确基因敲入,可提高基因敲入效率和精确性,减少修饰误差。
常用HDR模板及优缺点
(1)单链DNA(single-strand DNA, ssDNA)
ssDNA作为CRISPR基因敲入供体模板,由于其具有较简单的核苷酸结构、非特异性整合概率低、细胞毒性较低等特性,可实现准确CRISPR基因敲入,在HDR修复中被广泛使用。
优点:ssDNA在细菌和低等真核细胞中显示出较好重组效率;易于合成和转染到细胞中,且具有较高的HDR效率。ssDNA模板可用于点突变、小片段插入和删除等操作。
缺点:ssDNA插入的长度较短,不适用于大片段DNA的插入。
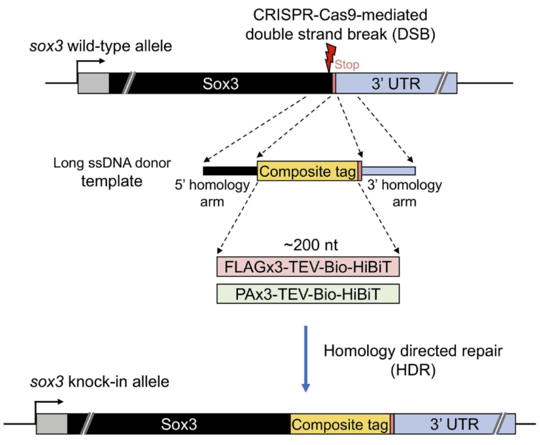
图2 使用ssDNA作为模板诱导CRISPR-Cas9介导的双链断裂后HDR机制(Deshani et al.,2021)
(2)双链DNA(double-stranded DNA, dsDNA)模板
dsDNA模板通常是通过PCR扩增、合成或从细胞中提取的DNA片段,通常包含目标序列及其周围的同源序列,以提高HDR修复的效率。
优点:dsDNA模板可插入较长的DNA片段,适用于需要大片段DNA插入的基因编辑。
缺点:合成和转染效率较低且其易被细胞中的核酸酶降解。dsDNA易被NHEJ修复途径合并,从而导致同源臂复制或dsDNA模板的部分整合。通过NHEJ同源独立性插入片段可能会发生双链断裂(double-strand break,DSB)脱靶或者产生内源性DSBs。此外,dsDNA模板还存在细胞毒性,线型或质粒dsDNA转染效率较低等短板。
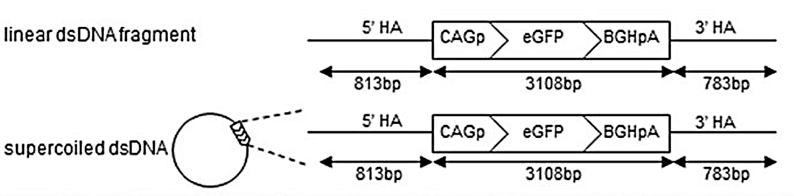
图3 使用长双链DNA模板的CRISPR/Cas9介导的HDR策略(Sachiko et al.,2019)
(3)RNA模板
RNA可作为HDR模板参与 CRISPR/Cpf1介导的DNA同源重组修复。
优点:与DNA模板相比,RNA模板具有易于合成优势。此外,RNA模板可在细胞内产生更高的修复效率,与RNA模板更易地被细胞摄取和转录成DNA,促进了同源重组修复的进程有关。同时RNA模板可避免DNA模板的不良影响(如细胞毒性、不良的基因重排和不必要的基因剪接事件等)。
缺点:由于RNA的易降解性和低稳定性,其在基因敲入中RNA模板的应用仍需进一步研究和优化。
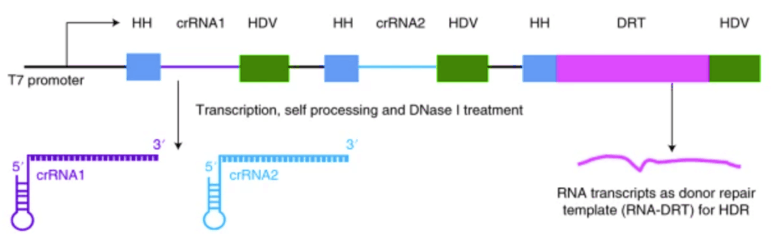
图4 crRNA和RNA DRT的制备示意图(Shaoya et al.,2019)
(4)基因组DNA
基因组DNA同样可作为CRISPR-Cas9介导的同源重组修复模板,用于实现精确的基因编辑。
优点:与其他类型的HDR模板相比,基因组DNA具有多个同源序列和大量的基因组信息,可用于实现较大范围的基因编辑,包括基因敲除、修复、敲入等操作。
缺点:基因组DNA通常较大,不同细胞类型和不同基因组区域的大小也不同,因此不确定性较高,导致使用基因组DNA作为HDR模板时,不同细胞或不同基因组区域的编辑效率和准确性不同。此外,基因组DNA含有大量的序列信息,可能包含多个同源序列或非同源序列,增加基因编辑的难度和不正确重组率;使用基因组DNA作为HDR模板涉及到人类基因组信息时需遵守相关的伦理和法律规定,以确保实验的安全和合法性。
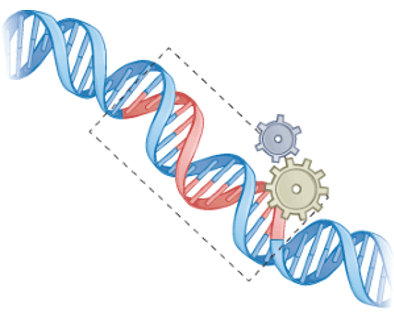
图5 CRISPR效应器(米色齿轮)与重组酶(灰色齿轮)结合,可与基因组等大序列DNA整合(Lei Tang et al.,2022)
在基因编辑技术的CRISPR/Cas体系中,基于同源介导修复 (homology directed repair, HDR) 机制开展的基因敲入是其重要应用之一,合适的HDR模板是决定基因敲入成败与效率的重要因素。
三、CRISPR基因敲入实验流程步骤
(以敲入RFP蛋白为例)
1、构建sgRNA-Cas9质粒及修复DNA模板质粒
(1)确定敲入位点
①若敲入的片段不参与翻译,则在想插入的附近设计sgRNA即可,并且根据sgRNA位置附近的序列设计donor载体(关于donor载体的设计在后文会详细介绍)。
②若敲入的基因参与翻译,并只是想让该蛋白表达蛋白,那么将外源基因插入到基因组中的safe harbor位点则是一个很好的选择。几乎所有的物种相应的针对其safe harbor的研究,人基因组的safe harbor有AAVS1,CCR5,以及ROSA26等。中国仓鼠的safe harbor有site A, T2,以及T9。需要注意的是将基因插到safe harbor处,需要一同准备promoter,enhancer等元件。
③若要敲入的基因参与翻译,并需要与宿主蛋白形成融合蛋白,那么需要考虑外源蛋白与宿主蛋白之间linker的问题,是使用刚性linker(rigid linker)还是柔性linker(rigid linker),还是选择自剪切多肽(2A Peptide),抑或是不使用linker。
(2)设计sgRNA
(设计sgRNA的具体步骤已在敲除篇中详细介绍,本文略)
(3)构建sgRNA-Cas9质粒
将设计好的sgRNA构建到PHB-pSpCas9(BB)-2A-Puro(PX459)载体上。

图6 PX459载体图
(4)设计donor质粒
①设计并制作两端同源臂(homologous arm):截取sgRNA靶点上游1000bp左右的序列,制作左同源臂(HA-L);截取sgRNA靶点下游1000bp左右的序列,制作右同源臂(HA-R)。
需要注意的是根据敲入位点,donor需要补齐一些序列,如需将RFP序列插入A基因终止密码子后而sgRNA设计的位置在终止密码前10个氨基酸处,那设计donor时需要将这10个氨基酸对应的碱基序列加在左侧同源臂上。此外,donor质粒上sgRNA序列需要进行同义突变,防止重复编辑。
②将需要敲入的基因序列(RFP序列)放置于左右同源臂之间,一起合成克隆于pcDNA3.1载体上。
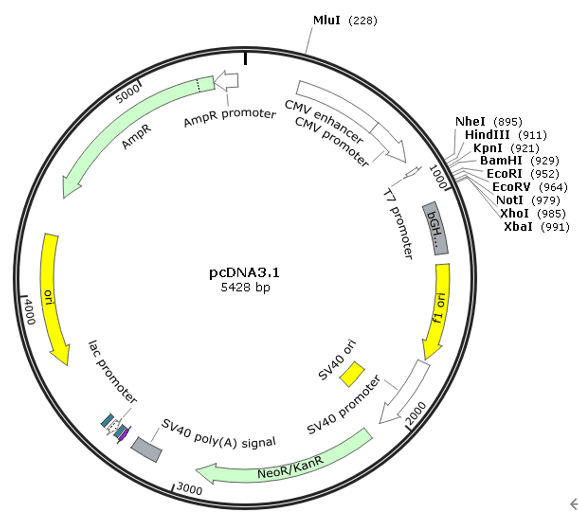
图7 pcDNA3.1载体图
2、将sgRNA-Cas9、修复DNA模版共转目的细胞
用lipofiter3.0转染试剂将上述两种质粒转染至目的细胞。
3、嘌呤筛选,检测荧光
本例子做的荧光蛋白敲入可以直接根据红色荧光信号进行分选。而其他的基因敲入需要根据gRNA质粒里面的荧光进行筛选或者进行puro抗性筛选,然后通过PCR等手段检测基因是否敲入。
4、PCR检测基因组
为了证实在基因组DNA中敲入RFP,设计靶向RFP上游的5’同源臂和靶向RFP下游的3’同源臂的一对引物。对照PCR产物大小即可知敲入是否成功。
5、单克隆分选
前期工作:准备好96孔板,在96孔板的四周加上300ul的DPBS(含双抗),中间6×10的孔中,加入300ul完全培养基(含双抗)。最后用无菌的parafilm封好四周。
(1)将转染结束的细胞消化下来
(2)500xg离心3min,用PBS重悬
(3)细胞悬液过300目细胞筛,筛出单细胞悬液
(4)加入PBS和培养基的96孔板,以及单细胞悬液一同置于冰上,等待流式分选
(5)上机分选,准备好的60个孔,每个孔分选入1个细胞
(6)分选后仍然将96孔板放在冰上保存
(7)将细胞放回培养箱中培养,5天后换液
(8)等待细胞分裂增殖到超过50%汇合度(一般会需要2周时间)
(9)消化细胞,随后利用孔板离心机离心细胞,弃消化液
(10)加入300 μl完全培养基到每个孔中,平均传到两块96孔板中(150 μl每孔),一块用于传代,一块用于PCR鉴定
(11)通过设计引物,进行PCR,检测基因是否敲除
(12)为进一步验证,将PCR产物送去Sanger测序
6、将敲入正确的多个单克隆进行扩增并冻存
需要注意的是尽可能多留几株不同的单克隆,以免某些单克隆的细胞特征与WT细胞差异过大。(一般文章中会使用3个不同的单克隆)
本期内容针对CRISPR-Cas9基因敲入系统(knock in)的实验原理、HDR模板的选择、及实验流程步骤等三大方面进行了详细介绍,希望对CRISPR-Cas9基因敲入系统研究方向的小伙伴可以有所裨益。下期我们将继续为大家介绍CRISPR-Cas9系统在转录调控中的应用,敬请关注!
参考文献:
[1] Murillo-Rodríguez E,Rocha B N,Veras B A, et al. The End of Snoring? Application of CRISPR/Cas9 Genome Editing for Sleep Disorders[J].Sleep and Vigilance, 2018,2(1).
[2] Sachiko Okamoto, et al. Highly efficient genome editing for single-base substitutions using optimized ssODNs with Cas9-RNPs. Sci Rep 2019, 4811.
[3] Deshani C. Ranawakage, et al. Efficient CRISPR-Cas9-Mediated Knock-In of Composite Tags in Zebrafish Using Long ssDNA as a Donor. Front Cell Dev Biol 2021,8:598634.
[4] Shaoya Li, et al. Precise gene replacement in rice by RNA transcript-templated homologous recombination. Nature Biotechnology 2019,37,445–450.
[5] Lei Tang, et al. Loading large DNA cargoes. Nature Methods 2023,20,33.8. Jinlin Wang, et al. Efficient targeted insertion of large DNA fragments without DNA donors. Nature Methods. 2022,19,331–340.









 7651
7651











