质粒“plasmid”一词由约书亚·莱德伯格(Joshua Lederberg)于1952年提出,被定义为染色体外遗传元件1。得益于其易用性、自我复制、稳定性,质粒已是现代生命科学研究中不可或缺的工具,无论是在基础研究还是转化应用领域,质粒的身影都随处可见,因此作为一个合格的生命科学科研人,质粒构建是一个非常基础的傍身技能。今天小编将带大家探一探质粒构建的玄机,后面几期内容大家还将陆续看细胞质粒转染、qpcr/wb检测基因表达等精彩内容。

闲言少叙,话归正题,说回质粒构建。
一、认识载体
知己知彼方能百战得胜,做质粒构建,自然要先了解质粒的组成。质粒的组成结构主要是复制起点(ORI)、筛选标记、多克隆位点、启动子。
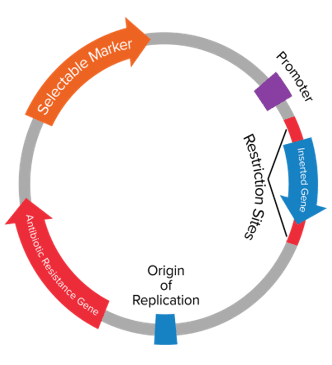

不同用途的质粒在结构上会有一些差异。克隆载体比较简单,一般仅有复制起点、筛选标记和克隆位点,而表达载体在克隆位点上游有启动子,下游有蛋白标签。原核表达载体经常还有操纵子元件,比如乳糖操纵子、阿拉伯糖操纵子。真核表达载体下游通常会有真和抗性、荧光蛋白标签,用于条件性调控的载体带有诱导响应元件,如tet、flox元件等。根据是直接转染表达还是包装成病毒颗粒后再感染细胞,又可以将其分为普通表达质粒和病毒质粒,病毒质粒包括慢病毒质粒、腺病毒质粒、腺相关病毒质粒等。
二、载体构建方法
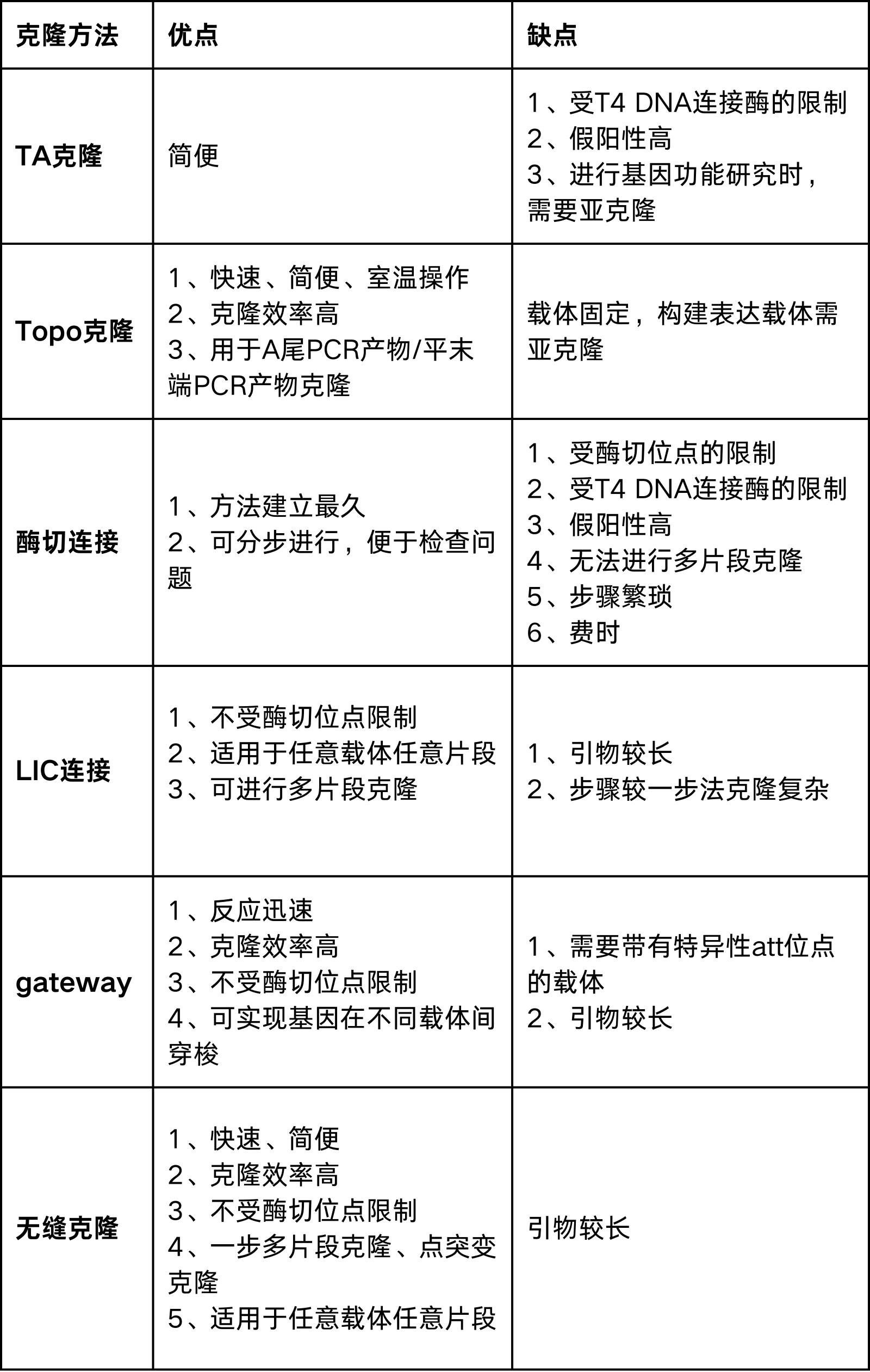
无论使用哪种载体,构建载体的中心思想就是把目的序列正确的插入到载体的多克隆位点中去。目前载体构建的方法也是多种多样,常用的有TA 克隆、酶切连接、LIC克隆、gateway和同源重组。
TA克隆
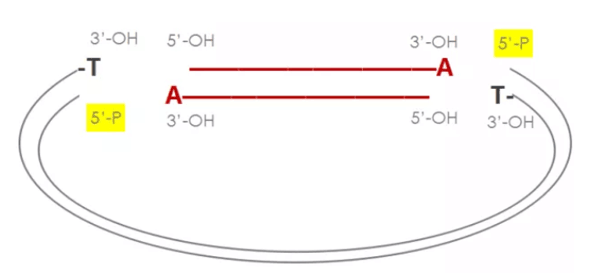
TA克隆技术(TAcloning)利用Taq聚合酶具有末端转移酶(TdT)活性,但却不具有3'-5'端外切酶校准活性的特点,可在PCR产物的3'端加上一个非模板依赖碱基“A”。而经过特殊处理的T载体带有3'端突出的碱基“T”,在连接酶的作用下,可与PCR产物3'末端的非模板依赖A连接,从而把PCR产物直接插入到质粒载体的多克隆位点中。TA克隆可以将不确定具体序列信息的PCR产物连接到T载体上,在进行序列确认和基因克隆中经常用到,比如在基因编辑中,分选编辑后的单克隆细胞时,就需要将编辑区域进行PCR扩增,将PCR产物做TA克隆后再进行测序分析,判断编辑结果是否满足需求。
酶切连接
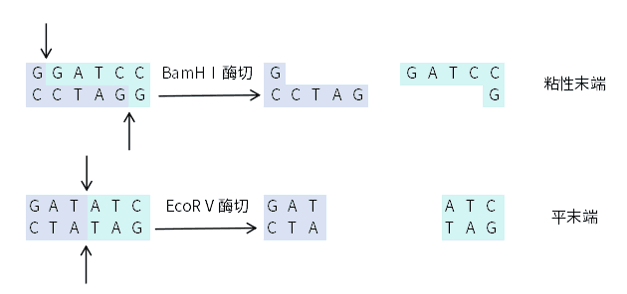
该方法可称是最经典的载体构建方法。它是用相同的限制性内切酶消化质粒和含有目的片段的DNA,得到具有相同的粘性末端或平末端的线性DNA,酶切产物进行纯化后,再利用DNA连接酶将线性化质粒与DNA片段共价结合,得到重组质粒。酶切连接实验中需要选择合适的酶切位点,酶切位点在载体和插入片段中需要单一,否则在酶切过程中会将载体和片段切成碎片。
LIC克隆
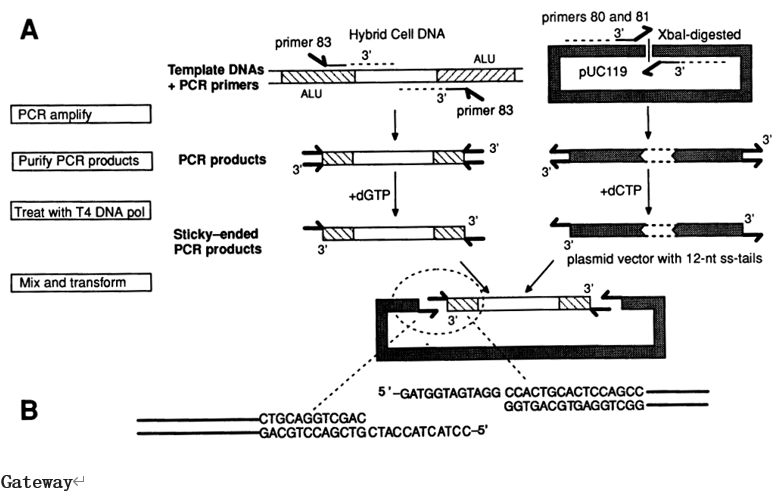
技术原理LIC技术全称为ligation–independent cloning(不依赖于连接反应克隆),在1990s首次由Aslanidis和de Jong提出(图1)2。LIC-PCR方法首次将同源重组引入体外基因克隆,利用了T4 DNA聚合酶的5'-3'聚合酶活性和3'-5'外切酶活性。外切酶活性使得载体和插入片段之间产生互补的突出端,聚合酶的活性利用dNTPs和互补DNA链模板恢复了DNA双链。在扩增插入片段的反应中掺入dGTP将核酸外切酶活性限制在第一个互补C残基上,T4 DNA聚合酶的聚合酶和外切酶活性在互补链第一次出现C时达到“平衡”。连接的片段有4个缺口,在转化过程中被大肠杆菌修复并产生环状质粒。
Li等人简化了这种方法,并于2007年开发了序列和连接非依赖性克隆SLIC(图2)[4]。使用SLIC时,克隆载体同样使用限制性内切酶或反向PCR线性化,而插入片段用PCR扩增,以引入与线性化载体互补的两个短同源末端。将插入片段和线性化载体混合并用T4 聚合酶处理,T4聚合酶发挥其3′-5′核酸外切酶活性,在同源末端形成单链区域。通过添加单个dNTP终止消化,然后在大肠杆菌中转化。
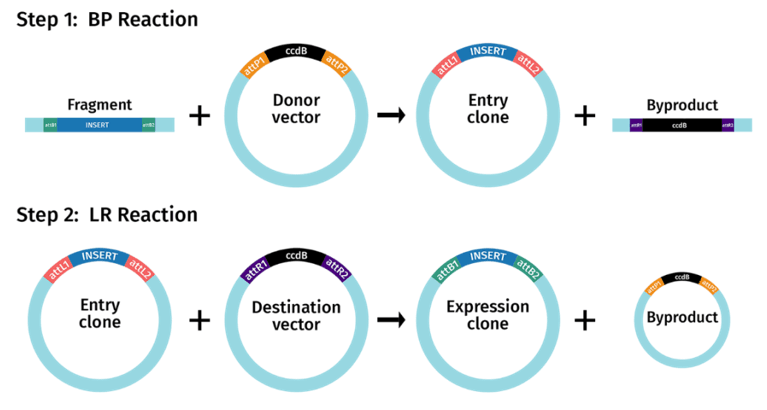
Gateway克隆技术是由Invitrogen公司利用Lambda噬菌体感染细菌时发生的整合和切除重组反应而开发的。在噬菌体和细菌的整合因子(INF、Int)的作用下,lambda的attP位点和大肠杆菌基因组的attB位点可以发生定点重组,lambda噬菌体DNA整合到大肠杆菌的基因组DNA中两侧产生两个新位点:attL和attR。相比于其他方法,gateway无需对载体进行线性化。
同源重组(Gibson assembly)
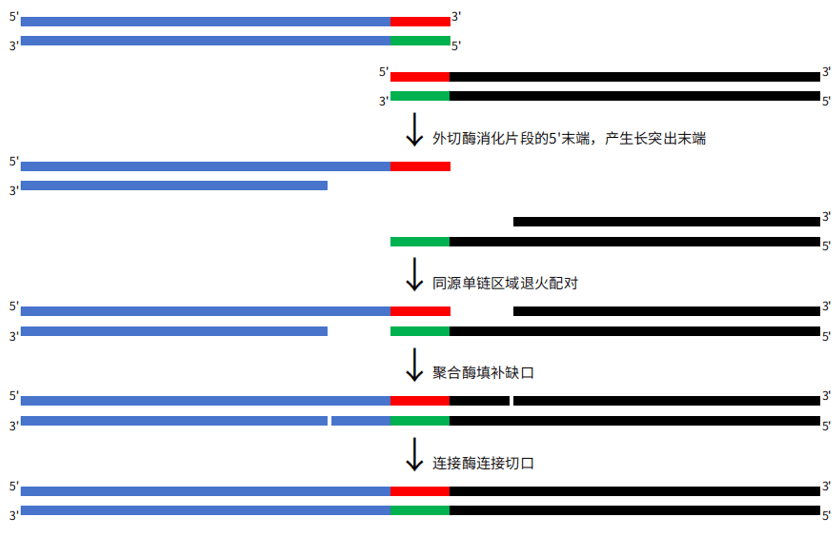
2009年J. Craig Venter研究所的Dr. Daniel Gibson及其同事提出Gibson assembly克隆技术3,4,Gibson连接时首先需要获得末端含有同源区域的DNA片段,一般是通过PCR获得,然后这些片段和酶混合物共孵育完成连接,该酶混合物含有三个不同的酶:核酸外切酶、聚合酶、连接酶。外切酶用于消化片段的5'末端,产生长突出末端,允许同源单链区域退火;聚合酶填补退火片段的缺口;连接酶活性能连接组装后的DNA切口。于是含有同源序列的两个或多个片段,在外切酶进行切割后,在同源序列处退火,而后在聚合酶和连接酶的作用下,产生一个完整的长链。
几种克隆方法优缺点比较
三、无缝克隆法构建载体
以上提到的这些载体构建方法中,同源重组因操作简便,成功率高,因此获得广泛应用,下面以汉恒生物的无缝克隆试剂盒为例介绍同源重组构建质粒的详细操作流程。
1、线性化载体制备
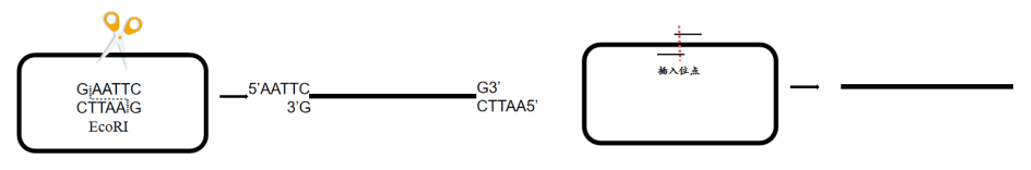
除了gateway技术外,其他构建方法均需要对载体进行线性化处理。载体线性化有酶切和反向PCR两种方式,在对载体进行线性化之前,首先要选择合适的克隆位点。尽量选择无重复序列且载体克隆位点上下游20bp区域内GC含量在40%-60%之间的位点进行克隆。
酶切制备线性化载体,既使用限制性内切酶将质粒切开,推荐使用双酶切方法使载体线性化完全,降低转化背景(假阳性克隆);如若使用单酶切线性化,可以在说明书推荐的时间基础上适当延长酶切时间以减少环状质粒残留,可有效降低转化背景。
采用反向PCR扩增制备线性化载体时,推荐使用高保真聚合酶进行扩增,可以有效避免扩增突变的引入。50μl的PCR体系中,使用0.1-1ng环状质粒模板,或使用预线性化质粒作为模板,这样做可以有效减少环状质粒模板残留,提高后续挑单克隆的阳性率。
2、插入片段制备
插入片段通过与线性化载体两端一致的15-25bp同源臂序列与线性化载体进行重组,同源臂通过pcr引物添加来引入。
(1)引物设计:在插入片段正反向扩增引物的5’端引入与线性化载体两末端同源序列,使扩增后的插入片段5'和3'最末端分别带有和线性化克隆载体两末端对应一致的同源序列(15-25 bp)。设计好的引物包括同源臂(15-25bp,GC含量40%-60%)+特异性引物。计算引物退火温度时,只需计算基因特异性引物的Tm值,引入的同源臂序列不参与计算。由于引入了同源臂序列,引物通常会超过40bp,在引物合成时推荐选用PAGE纯化方式,可以提高阳性克隆率。
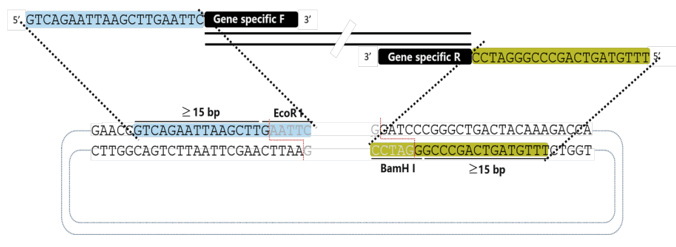
(2)插入片段PCR扩增要使用高保真聚合酶扩增,以避免在扩增过程中引入突变。为了提高pcr扩增效率,可以先设置梯度PCR确定最佳退火温度。按照所使用的高保真聚合酶的特性配置PCR体系,根据扩增片段的大小设置合适的延伸时间,确定最佳的PCR条件。
(3)通过琼脂糖凝胶电泳回收线性化的载体和pcr扩增的目的基因片段。回收产物测定好核酸浓度后就可以开始下一个步骤了。
3、连接反应
汉恒生物无缝克隆试剂盒20ul重组反应体系中核酸(线性化载体+PCR产物)使用量为0.02-0.5 pmol,载体与插入片段的摩尔必为1:2-1:5。如果插入片段长度大于克隆载体时,则需要将插入片段看作是载体,而将载体看作是插入片段来计算需要加入的核酸片段量。如果线性化克隆载体和插入片段扩增产物未进行DNA纯化直接使用,那加入总体积应不超过反应体系体积的1/5(4ul);相反,如果回收的DNA浓太高则需要做稀释,保证各组分加入体积不低于1ul。
冰上配置反应体系:
根据自身需要,可以按照说明书配制20ul体系,也可以配制10ul体系(10ul体系各成分减半,分子实验老炮常这样干),配好的反应体系置于50℃孵育 20min。
4、转化连接产物
(1)冰上解冻感受态细胞(如:DH5α Competent cell),取 2ul重组产物加入融化好的感受态细胞中,轻轻混匀,不能震荡混匀,将该混合物置于冰上 30 min。
(2)轻轻摇匀后放入 42℃水浴中 1~2 min 进行热激,然后迅速放回冰中,静置 3~5min。
(3)在超净工作台中向上述各管中分别加入500μl LB 培养基(不含抗菌素)轻轻混匀,然后固定到摇床的弹簧架上 37℃震荡 1h,转速 250 rpm。
(4)在超净工作台中取上述转化混合液100~300μl,分别滴到含相应抗生素的固体 LB平板培养皿中,用无菌涂布棒涂布均匀,37℃培养箱中培养过夜。
5、菌落PCR验证
感受态转化后一般16h便可长成单克隆,待平板上单克隆长成后便可以进行挑单克隆验证。
(1)每个平板上挑取单克隆若干个进行菌落PCR鉴定。用5ul吸头挑取菌斑到2ml EP管中(装有0.5-lml含相应抗生素的LB培养基),反复吸打几次后吸1-2ul菌液加入PCR反应体系中作为扩增模板,并做好标记,并将EP管于摇床中培养(27℃,250rpm)。
(2)PCR体系扩增引物可以选择选择载体骨架上的引物或目的基因特异引物,也可用一个载体引物与一个目的序列特异引物相组合,根据扩增产物的大小和使用的酶确定延伸时间和PCR程序。琼脂糖凝胶电泳检测克隆正确时应有于理论序列片段大小一致的条带出现。
(3)将菌落PCR鉴定为阳性的菌落继续37℃、250rpm培养过液,阴性克隆直接灭菌处理。
(4)阳性克隆sanger测序验证。菌落pcr可验证目的片段是否插入了载体中,但是不能验证插入序列是否发生了突变,所以需要sanger测序进一步确认序列。
6、保存与复苏
保存测序正确的单克隆。一般采用冻存甘油菌的方式保存阳性克隆,向菌液中加入无菌甘油溶液,使甘油终浓度在20-30%,混匀后直接液氮速冻后-80℃保存。
再次使用时建议先对冻存甘油菌进行复苏。将甘油菌室温溶解后用接种环蘸取少量菌液在含相应抗生素的LB平板上做划线梯度稀释,37℃培养过夜后挑去长出的单克隆培养、抽提质粒。
四、载体构建常见问题
1、PCR扩增目的基因时,琼脂糖凝胶电泳有多个条带
产物有多个条带可能有一下原因:1、设计的引物有特异性不好,2、引物添加的接头过长(同源重组和gataway),引物之间形成引物二聚体,3、pcr扩增的退火温度过低,导致错配率高。
解决方案:如果多条产物中目的条带清晰,可以考虑直接切胶回收,如无法分清目的条带目明显,可做如下1、扩增目的基因的引物位置固定,若出现杂带可以适当延长引物序列长度,2、引物接头过长可以适当减少引物用量,或者可以分两次添加接头序列上去,3、适当提高退火温度(会降低产物得率)。
2、转化后平板上长出的单菌落检测后假阳性很高
假阳性可能原因有1、载体酶切不完全,2、培养基抗生素失效,3、载体发生自连
对应解决方案:1、可以适当延长酶切时间,酶切产物进行琼脂糖凝胶电泳回收纯化;2、抗生素失效平板上的菌落会生长密集、甚至成片,可以在涂板时加涂一个未转染过的感受态加已排除,如确认时抗性失效,及时更换抗生素;3、载体自连发生在单酶切时,可以通过对线性化载体去磷酸化和使用双酶切的方式加以避免(双酶切时要先择粘性末端不同源的限制性内切酶)。
3、转化后未长出克隆
转化未长出克隆可能是连接不成功,或者连接成功但转化不成功。出现这些情况可能有以下原因:
1、引物设计错误:检查插入片段添加的同源臂序列与载体上的同源臂序列是否对应。
2、载体与目的片段使用比例失调。根据载体和插入片段碱基数计算调整二者加入量。
3、载体与目的片段不纯。对线性化载体、PCR产物进行切胶回收纯化。
4、操作问题或试剂盒失效。建议做试剂盒附带的阳性对照实验排除操作原因。
5、感受态细胞效率低。建议用转化效率>107 cfu/μg的感受态细胞。
6、此外抗生素使用错误即使来连接与转化都没有问题,平板上叶不能长出相应克隆,所以确保抗生素的正确使用。
4、PCR扩增目的片段经常出现突变
1、选择高保真酶进行扩增,可以有效避免扩增过程中碱基的突变。2、核对引物序列,避免因为引物序列错误而引入突变。3、循环数越高出现突变的几率越高,扩增循环数不要太高,一般不超过30个扩增循环。
5、保存的菌液直接摇菌摇不起来。
冻存菌液可能活力比较低,直接吸取菌液摇菌扩增不起来,可以将冻存菌液现在平板上划线活化后再挑菌斑摇菌。
五、结语
本期干货介绍了实验室常见的载体构建方法、无缝克隆构建载体的详细操作步骤以及载体构建的注意事项,希望对大家有所帮助。汉恒生物可提供多种类型载体构建服务,大家可通过汉恒生物微信公众号或拨打官网技术服务热线(400-092-0065)进行咨询。下期干货将继续为大家介绍如何将质粒高效转染到目的细胞,欢迎大家持续关注~
六、参考文献
1、Wein T, Dagan T. Plasmid evolution. Curr Biol. 2020 Oct 5;30(19):R1158-R1163. doi: 10.1016/j.cub.2020.07.003.
2、Aslanidis C, de Jong PJ. Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 1990 Oct 25;18(20):6069-74. doi: 10.1093/nar/18.20.6069. PMID: 2235490; PMCID: PMC332407.
3、Gibson DG, Benders GA, Andrews-Pfannkoch C, Denisova EA, Baden-Tillson H, Zaveri J, Stockwell TB, Brownley A, Thomas DW, Algire MA, Merryman C, Young L, Noskov VN, Glass JI, Venter JC, Hutchison CA 3rd, Smith HO. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science. 2008 Feb 29;319(5867):1215-20. doi: 10.1126/science.1151721. Epub 2008 Jan 24. PMID: 18218864.
4、Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009 May;6(5):343-5. doi: 10.1038/nmeth.1318. Epub 2009 Apr 12. PMID: 19363495.









 3227
3227










