肥胖(Obesity)是体内脂肪积聚过多而呈现的一种状态,其增加了代谢性疾病、心血管疾病甚至癌症等疾病发生的风险,对人类健康产生了巨大的威胁。生酮饮食(Ketogenic Diet,KD)是一种高脂肪、蛋白质适中和极低碳水化合物(或无碳水化合物)的饮食,在这种饮食条件下,机体的主要供能方式由葡萄糖代谢转变为脂肪代谢,由脂肪在肝脏中产生的酮体为全身供能。这种饮食方式被证明可以有效替代饮食限制来进行减肥,并且得到广泛推广,但其具体的机制尚不清楚。
生长分化因子15(GDF15)是转化生长因子β(TGF-β)超家族的成员,在20世纪90年代末被首次发现。研究显示,在致肥胖条件下通过转基因过表达GDF15或重组GDF15给药可改善代谢参数,促使其成为预防和治疗肥胖的新靶点,并且GDF15的一个特征是抑制食物摄入,而KD饮食的动物食物摄入量减少,表明GDF15与KD的肥胖管理之间存在潜在联系。
2023年12月5日,西北农林科技大学吴江维团队和西京医院李晓苗团队联合在Cell Metabolism上在线发表了题为“GDF15 is a major determinant of ketogenic diet induced weight loss”的研究论文。该研究发现KD饮食可以抑制人、猪和小鼠的能量摄入和减轻其体脂肪量,诱导血浆GDF15的轻微升高。并且,作者通过使用GDF15缺陷型(GDF15-/-)小鼠以及GDF15受体GFRAL缺陷型(GFRAL-/-)小鼠为实验动物,发现了GDF15-GFRAL信号轴对KD的减重作用至关重要。同时通过用AAV8-GDF15-shRNA和AAV8-PPARγ-shRNA敲低小鼠肝脏中GDF15和PPARγ的表达,消除了KD对肥胖的管理作用,进而证实了KD诱导的GDF15来源于肝脏PPARγ的激活。另外,在肝脏PPARγ敲除小鼠中,血浆GDF15水平降低,KD饮食对肥胖管理的有益作用大大减弱,而注射AAV8-GDF15-overexpression或重组GDF15蛋白可以恢复KD对肥胖的管理作用。研究结果首次揭示了GDF15在KD饮食管理体重中发挥着不可或缺的作用,增加了对KD饮食发挥代谢调控作用的理解。
值得注意的是,在本研究中,作者使用了汉恒生物提供的AAV8-GDF15-shRNA和AAV8-PPARγ-shRNA病毒,成功敲低了小鼠肝脏中GDF15和PPARγ的表达。
下面,我们一起来了解具体的研究内容:
为了探究KD饮食对于肥胖管理的作用,作者首先给肥胖的小鼠进行KD饮食或者高脂(HFD)饮食15天,结果发现从第6天开始KD组体重逐渐下降,而HFD组体重逐渐升高;并且从第3天开始,KD小鼠的累计能量摄入(kcal)减少了16.48%,血浆GDF15增加了2倍。为了进一步验证KD是否特异性诱导GDF15的表达,作者给予小鼠几种常见的肥胖管理饮食,同时又设计了两种禁食方案:每隔一天禁食和限时禁食,并测量小鼠血浆中GDF15的浓度,结果显示,这些类型的饮食在测试时间内并不能提高小鼠血浆GDF15的水平。为了进一步验证KD与GDF15的联系,作者以小型猪为实验动物,进行KD饮食。研究结果显示,在KD饮食处理15天后,KD处理组的猪体重下降,血浆GDF15水平升高;此外,作者招募了一组肥胖患者进行为期2周的KD干预,研究结果显示,在干预结束后,患者的体重显著降低,并且血浆GDF15水平显著升高。这些结果表明KD饮食降低了体重,增加了血浆GDF15的水平。
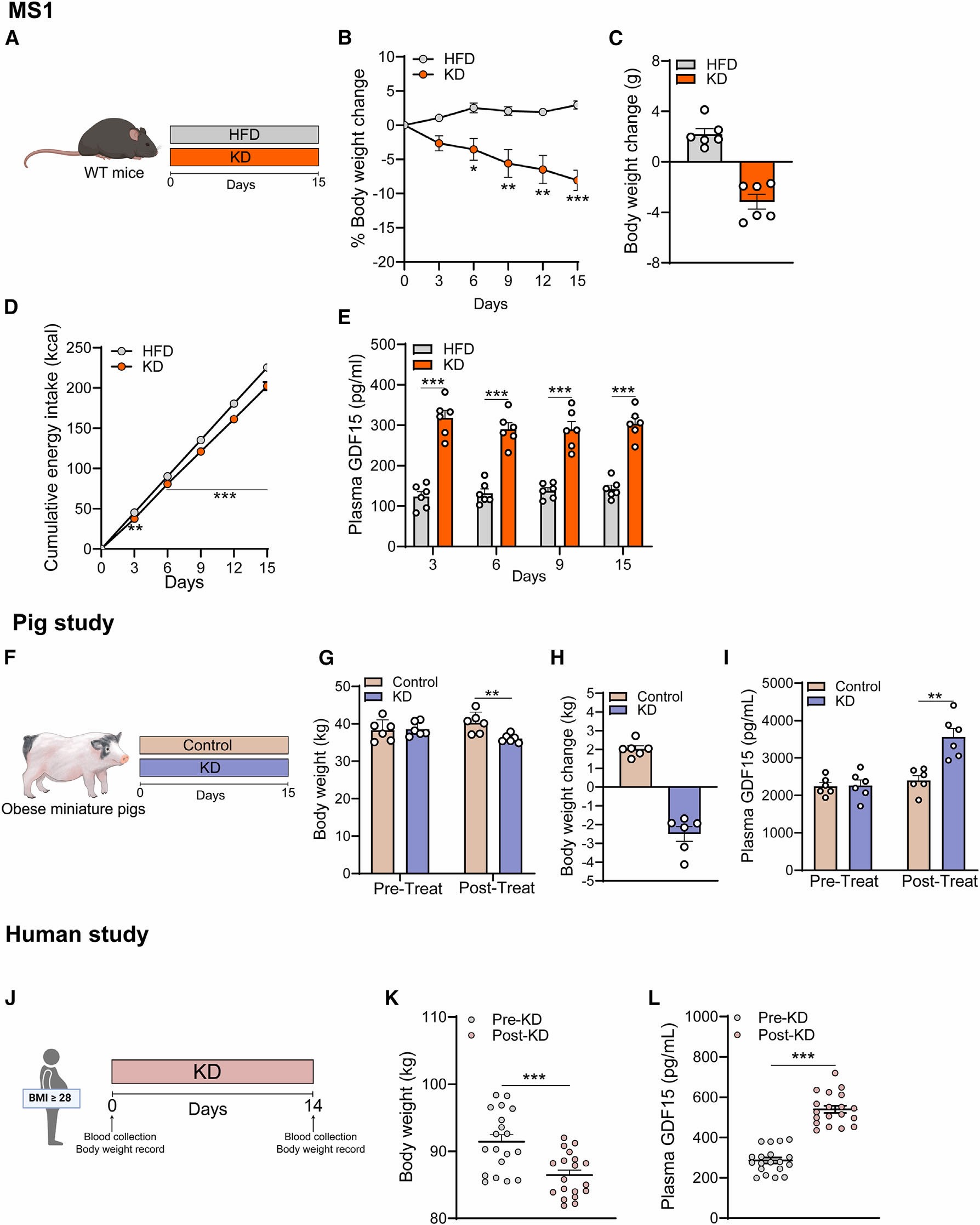
图1 KD饮食减轻体重,升高血浆GDF15的水平
接下来,为了探究GDF15在KD饮食减轻体重中发挥的作用,作者对GDF15缺陷型(GDF15-/-)小鼠和野生型(WT)小鼠同时进行30天的KD饮食或HFD饮食处理,研究结果显示,30天的KD饮食显著增加了WT小鼠血浆GDF15的水平,而GDF15-/-小鼠中血浆GDF15水平没有变化;同时,与HFD饮食(对照组)比较,KD饮食降低了WT小鼠的累计能量摄入量和体重,对GDF15-/-小鼠没有影响;但是,与WT小鼠比较,KD饮食反而增加了GDF15-/-小鼠的体重。与对照组相比,KD饮食降低了WT小鼠的脂肪、肝脏质量以及血浆甘油三酯(TG)、谷丙转氨酶(ALT)、天冬氨酸转氨酶(AST)和肝脏中TG含量,但GDF15-/-小鼠中没有发现类似的结果。此外,与对照组相比,KD饮食改善了WT小鼠胰岛素敏感性以及葡萄糖耐量,而GDF15-/-小鼠并没有表现出这些变化。这些结果表明GDF15对于KD饮食介导的肥胖管理是不可或缺的。
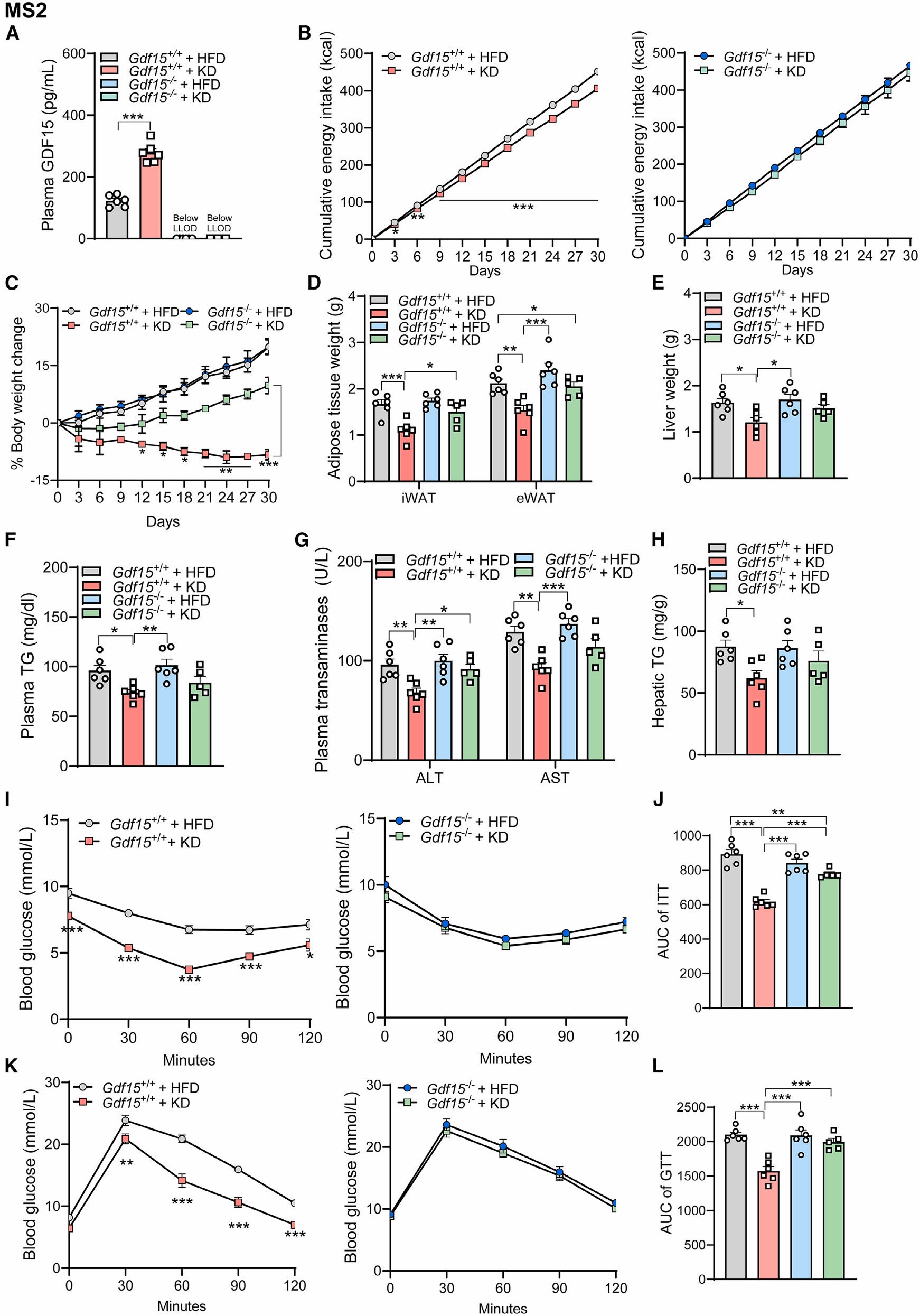
图2. GDF15对于KD饮食减轻体重是必需的
之后,为了进一步验证血浆GDF15的升高是否有助于KD饮食对肥胖管理的效果,作者用外源性重组GDF15蛋白干预小鼠,7天后将GDF15干预的小鼠分成2组,一组接受IgG处理,一组接受GDF15中和抗体处理。研究结果显示:GDF15中和抗体降低了WT小鼠的累计能量摄入量和体重。并且,与HFD饮食(对照组)比较,IgG处理的KD饮食小鼠体重减轻,而利用GDF15中和抗体处理后,KD饮食小鼠的体重开始荒蛮上升,这表明,GDF15直接介导了KD饮食引起的减重效应。
此外,作者在GDF15受体GFRAL缺陷型(GFRAL-/-)小鼠中也进行了相同的实验,研究结果显示:不管在野生型小鼠还是在GFRAL-/-小鼠中,KD饮食均会增加血浆GDF15的水平,但是在GFRAL-/-小鼠中,KD饮食对小鼠的累计能量摄入量、体重、脂肪质量、肝脏质量、血浆ALT、血浆AST和肝脏中TG含量均无明显影响;此外,在GFRAL-/-小鼠中,KD饮食对小鼠葡萄糖耐量和胰岛素敏感性的改善作用也未体现。这些结果表明GDF15-GFRAL信号在小鼠KD饮食的有益代谢作用中起着重要作用。
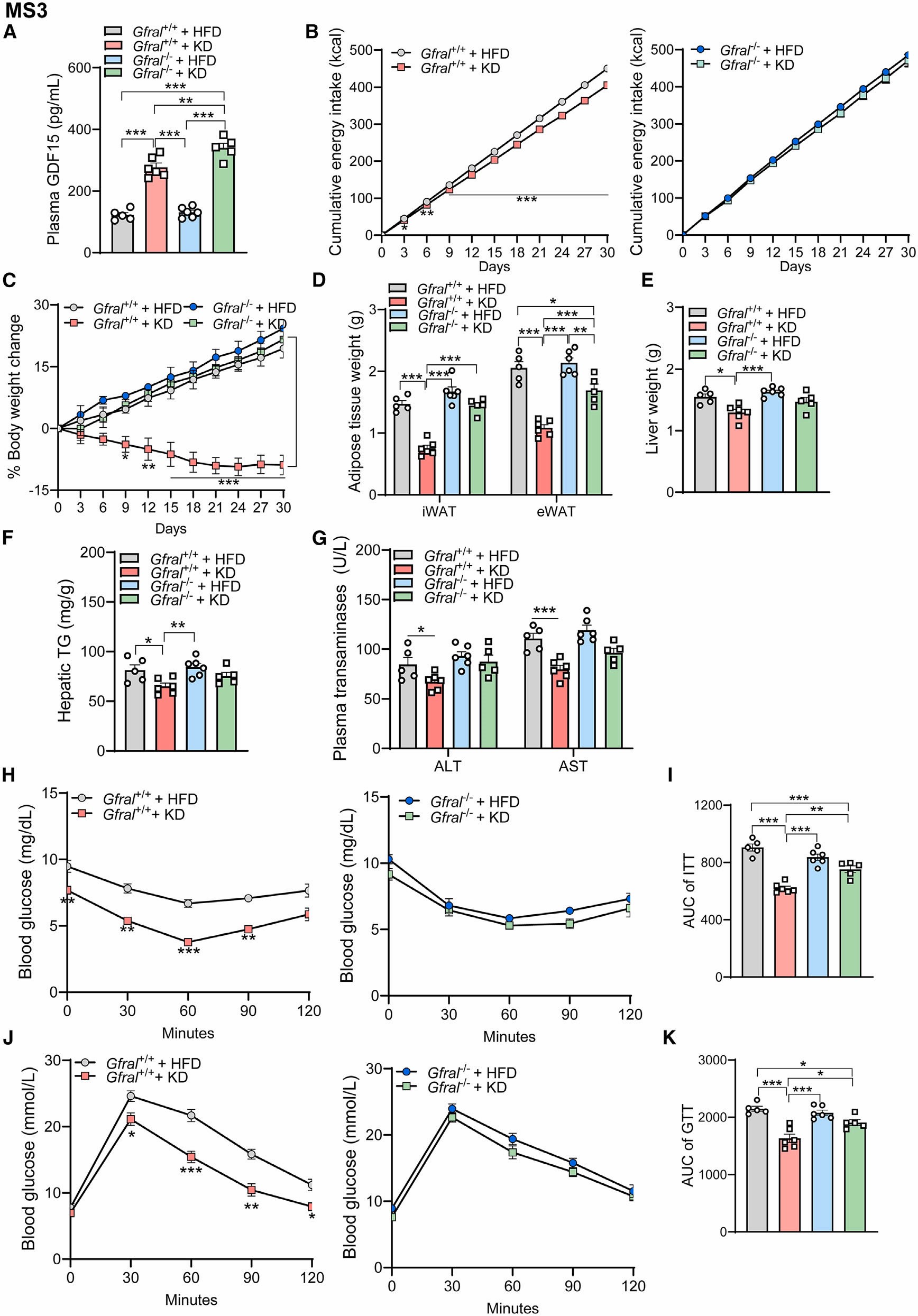
图3 GFRAL对于KD饮食减轻体重是必需的
为了测试能量摄入的减少或能量消耗的改变是否有助于KD饮食管理肥胖,作者对小鼠按能量进行配对饮食,研究结果显示配对饮食在很大程度上消除了KD饮食和HFD饮食小鼠之间的体重差异。接着,作者对KD饮食和HFD饮食的GDF15-/-小鼠和WT小鼠使用间接测热法,以确定能量消耗是否存在额外的影响。通过协方差分析(ANCOVA),以瘦体重为协变量,分析结果显示,KD饮食的小鼠无论GFD15是否存在,其能量消耗都高于HFD对照组。以上实验结果表明,KD饮食介导的肥胖管理既包括GDF15调节的能量摄入,也包括独立于GDF15的能量消耗。
另外,有研究报道,KD饮食可增加小鼠血浆FGF21的水平,这是一种已知的可增加能量消耗的因素。因此,作者测量了血浆中FGF21的水平,发现KD饮食的WT小鼠和GDF15-/-小鼠的FGF21水平升高,其肝脏中FGF21 mRNA水平也升高。为了探究KD饮食下独立于GDF15的能量消耗是否与FGF21有关,作者用FGF21中和抗体处理KD饮食的GDF15-/-小鼠和WT小鼠,结果显示HFD饮食和KD饮食的小鼠之间的能量消耗不存在差异,结果表明FGF21是KD饮食下能量消耗增加的原因。并且,FGF21中和抗体能消除HFD饮食的GDF15-/-小鼠和KD饮食的GDF15-/-小鼠的体重差异。这些实验结果表明KD饮食的肥胖管理的作用是通过协调GDF15介导的能量摄入抑制和FGF21介导的能量消耗而实现的。
之后,为了探究KD饮食诱导的GDF15主要来源于哪个组织,作者检测了肝脏、肾脏、心脏、骨骼肌、回肠和结肠组织中GDF15基因的表达水平。与对照组比较,KD干预的小鼠肝脏中GDF15 mRNA和蛋白的水平显著增加(同时,KD饮食的猪肝脏中GDF15 mRNA的表达明显增加);为了进一步探究KD是否能在体内诱导肝脏GDF15的产生,作者使用AAV8-GDF15(AAV8-GDF15-shRNA)敲低小鼠肝脏中GDF15的表达,研究结果显示,KD饮食不能显著增加AAV8-GDF15小鼠血浆中GDF15的水平,表明HD饮食诱导了肝脏GDF15的产生;并且,肝脏GDF15下调后,KD饮食对能量摄入的抑制作用、体重减轻和减脂效果明显降低。此外,在AAV8-GDF15小鼠中,KD对葡萄糖耐量和胰岛素敏感性的改善作用消失,即KD饮食和HFD饮食小鼠表现出相当水平的葡萄糖耐量和胰岛素敏感性。以上结果表明,肝脏是KD饮食诱导GDF15产生的主要部位。
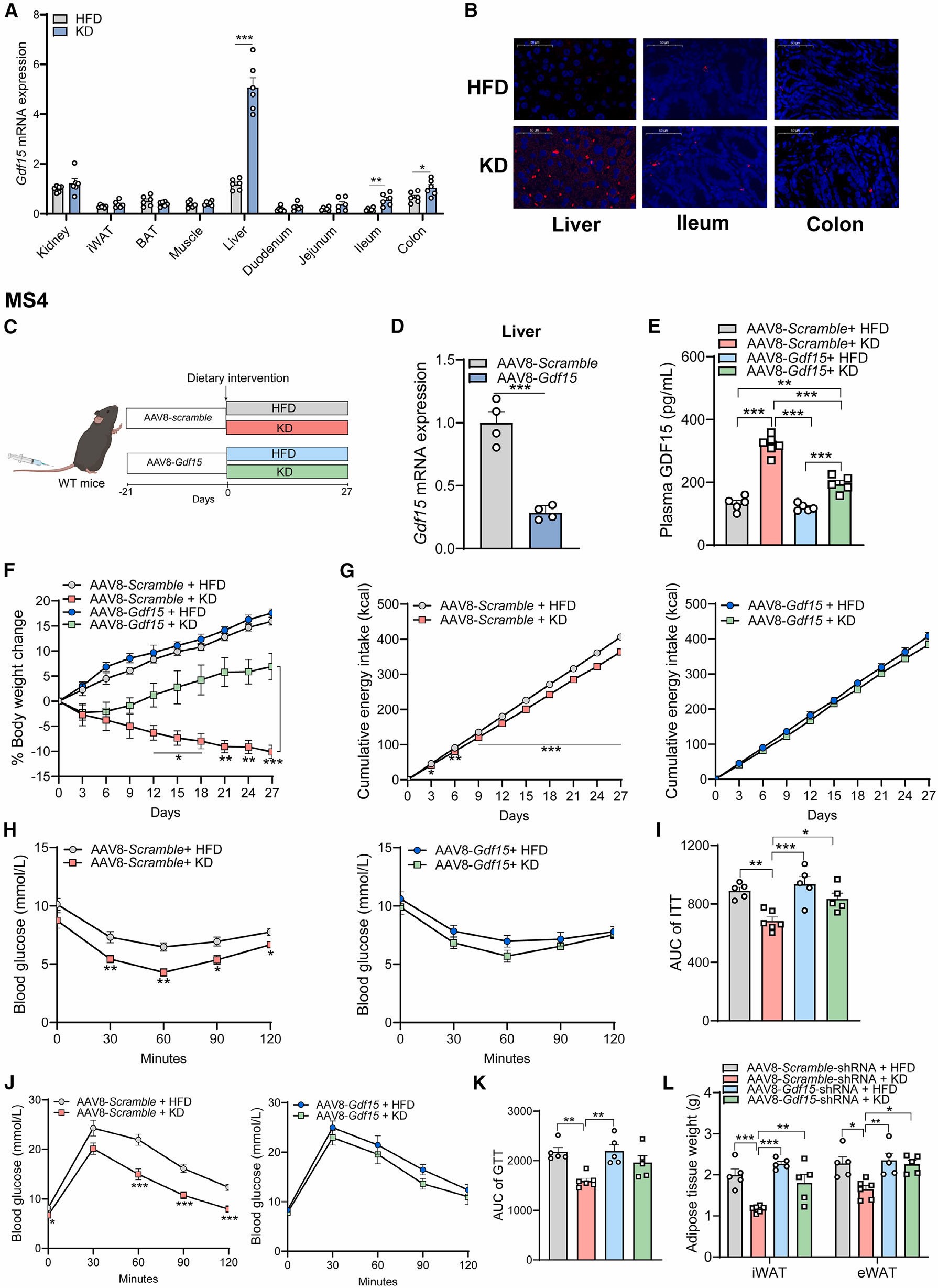
图4.KD饮食诱导的血浆GDF15主要来源于肝脏
接着,为了进一步探究KD饮食诱导GDF15的分子机制,作者对HFD和KD饮食的小鼠肝脏做了RNA-seq分析。KEGG通路分析显示,差异表达基因在多种信号通路中富集,其中包括PPAR信号通路及其靶基因在内的15个基因(Pparg、Cd36、Pltp、Cyp4a14、Slc27a1、Me1、Scd1、Cyp8b1、Cyp4a10、Cyp4a12b、Cpt1b、Acaa1b、Ehhadh、Acox1、Cyp4a12a)在KD饮食后显著上调,并且qPCR结果分析也证实了以上结果。接着,对小鼠肝脏中PPAR的成员(PPAR是核受体亚家族,有三个成员:PPARɑ、PPARγ和PPARβ/δ)进行检测,发现只有PPARγ mRNA和PPARγ蛋白的表达显著增加。并且,跟对照组比较,KD饮食的猪肝脏中PPARγ mRNA也显著增加。这些结果表明,PPARγ和GDF15之间可能存在相关性。之后,作者对GDF15启动子序列进行分析,筛选到4个可能的PPAR响应元件,并且双荧光素酶报告基因检测实验显示在小鼠正常肝细胞AML12中,PPARγ的表达能够激活GDF15启动子的转录活性。Chip-qPCR和双荧光素酶报告基因检测实验结果表明,PPARγ作为转录因子通过与GDF15启动子结合促进GDF15的转录和表达。为了进一步确定肝细胞是否能够响应PPARγ并促进GDF15的表达,作者用PPARγ的激动剂罗格列酮培养小鼠正常肝细胞AML12,结果显示,AML12细胞经过罗格列酮处理后,细胞中GDF15 mRNA表达升高,同时细胞上清中GDF15的水平也升高;并且,在AML12细胞中过表达PPARγ,也出现了类似的结果。同样,在人原代肝细胞中,罗格列酮处理或者PPARγ过表达均显著增加了细胞中GDF15 mRNA表达升高和细胞上清中GDF15的水平。此外,鉴于PPARγ在脂肪细胞中高度表达,作者进一步研究了脂肪PPARγ是否以与肝脏相似的方式发挥作用。作者对脂肪细胞3T3-L1进行了相同的处理(罗格列酮处理或过表达PPARγ),然而并没有检测到类似肝细胞或上清中GDF15的表达变化,进而排除了脂肪细胞中GDF15转录和产生的类似调节机制。此外,有研究表明肝脏整合应激反应(integrated stress response ISR)上调GDF15的表达。因此,作者通过检测ISR的关键转录调控因子ATF4和CHOP的肝脏蛋白水平进而研究KD饮食诱导的GDF15表达是否与ISR激活有关,结果显示,KD饮食小鼠中ATF4和CHOP的蛋白水平和HFD对照中的水平相似,进而排除了ISR在肝脏GDF15产生中的潜在作用。这些实验结果表明PPARγ以肝细胞特异性的方式上调GDF15的表达。
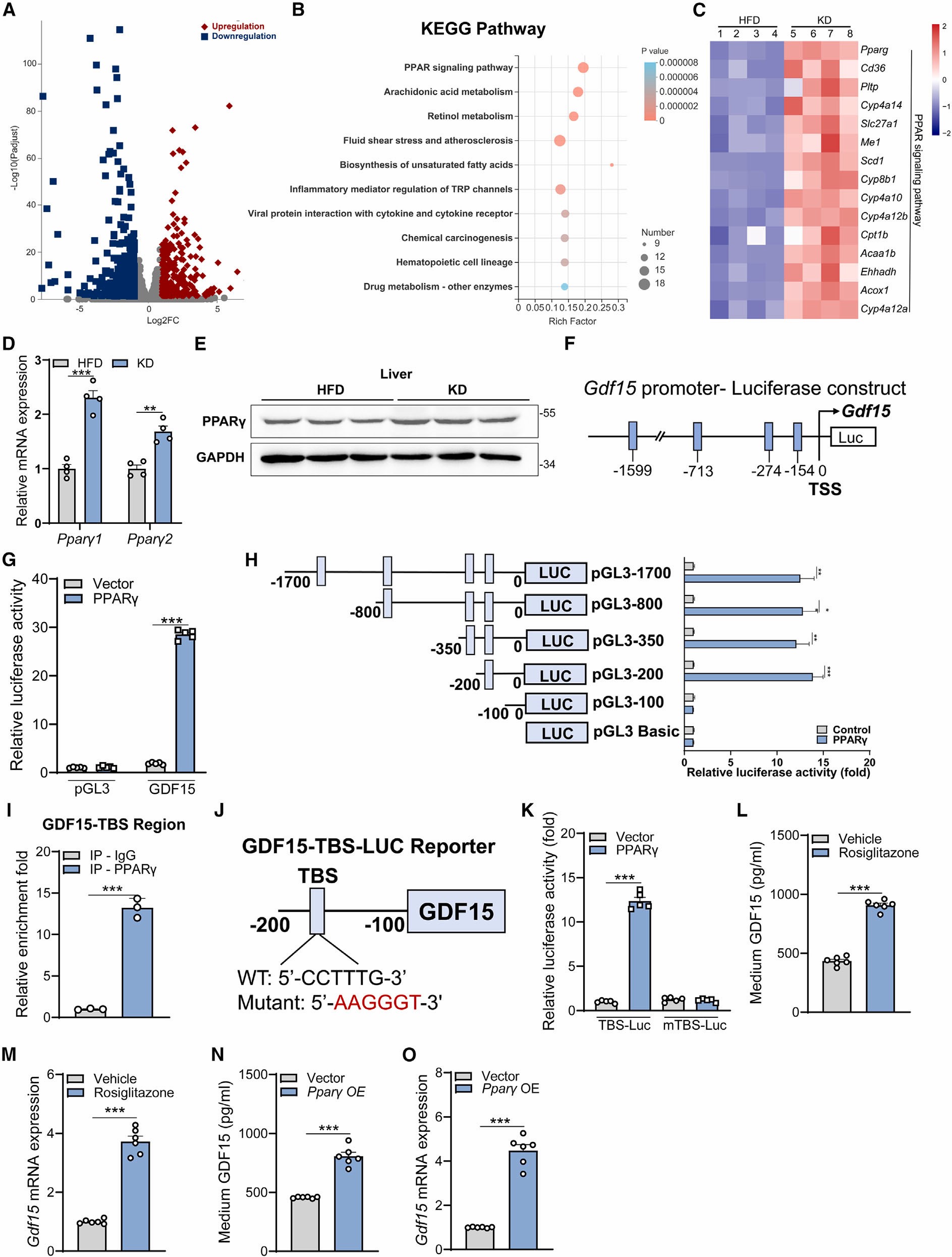
图5 肝脏而非脂肪组织中的Pparγ上调GDF15的表达
为了进一步探究PPARγ和GDF15在肝脏中的联系,作者使用AAV8-PPARγ-shRNA敲低小鼠肝脏中PPARγ的表达。与AAV8-scramble对照组比较,AAV8-PPARγ-shRNA小鼠经过KD饮食后,肝脏GDF15 mRNA的表达和分泌明显减少;并且肝脏PPARγ基因敲低后,KD饮食对体重管理的作用也明显减弱。此外,作者在肝脏特异性PPARγ基因敲除(PPARγ△Hep)小鼠中也测试了KD饮食对体重管理的作用,结果显示,KD饮食能诱导WT小鼠分泌GDF15,但在PPARγ△Hep小鼠中没有发现类似的效果,并且,在PPARγ△Hep小鼠中,KD饮食对能量摄入的抑制作用和脂肪量的减少效应消失。相同的,KD饮食显著降低了WT小鼠血浆TG、肝脏脂质含量和肝脏重量,而在PPARγ△Hep小鼠中没有发现类似的变化。此外,随着肝脏中PPARγ基因的缺失,KD饮食对葡萄糖耐量和胰岛素敏感性的改善作用也随之消失。以上实验结果表明,肝脏PPARγ是KD饮食对肥胖管理的关键分子。
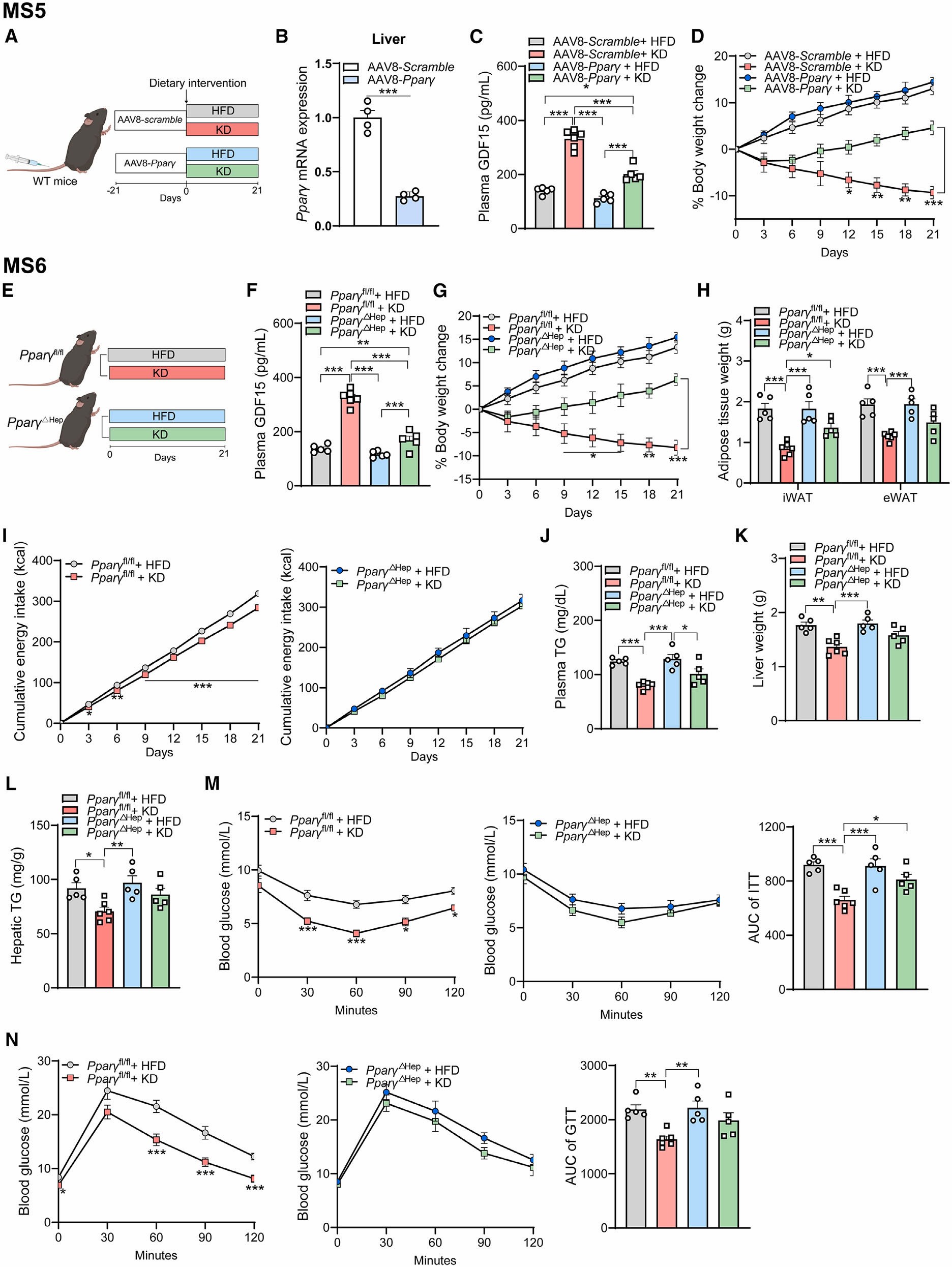
图6 肝脏PPARγ缺失小鼠血浆GDF15水平降低,且KD饮食对肥胖管理的效果消失
最后,为了探究KD饮食在PPARγ△Hep小鼠体内肥胖管理作用的消失是否是因为肝脏GDF15表达和血浆GDF15水平的下降引起的,作者进行了挽救实验,利用AAV8-GDF15-overexpression(AAV8-GDF15-OE)在肝脏中特异性过表达GDF15。与对照组(注射AAV Vector)比较,肝脏过表达GDF15可以显著增加小鼠血浆GDF15的水平,并且恢复了KD饮食对PPARγ△Hep小鼠肥胖管理的作用(抑制能量摄入、减轻体重、减轻脂肪量、恢复PPARγ△Hep小鼠葡萄糖耐量、降低肝脏TG含量)。随后,作者进一步通过给小鼠注射重组GDF15蛋白,观察GDF15是否能恢复KD饮食对PPARγ△Hep小鼠的肥胖管理作用。结果显示重组GDF15能有效地恢复KD饮食对PPARγ△Hep小鼠肥胖管理的作用。
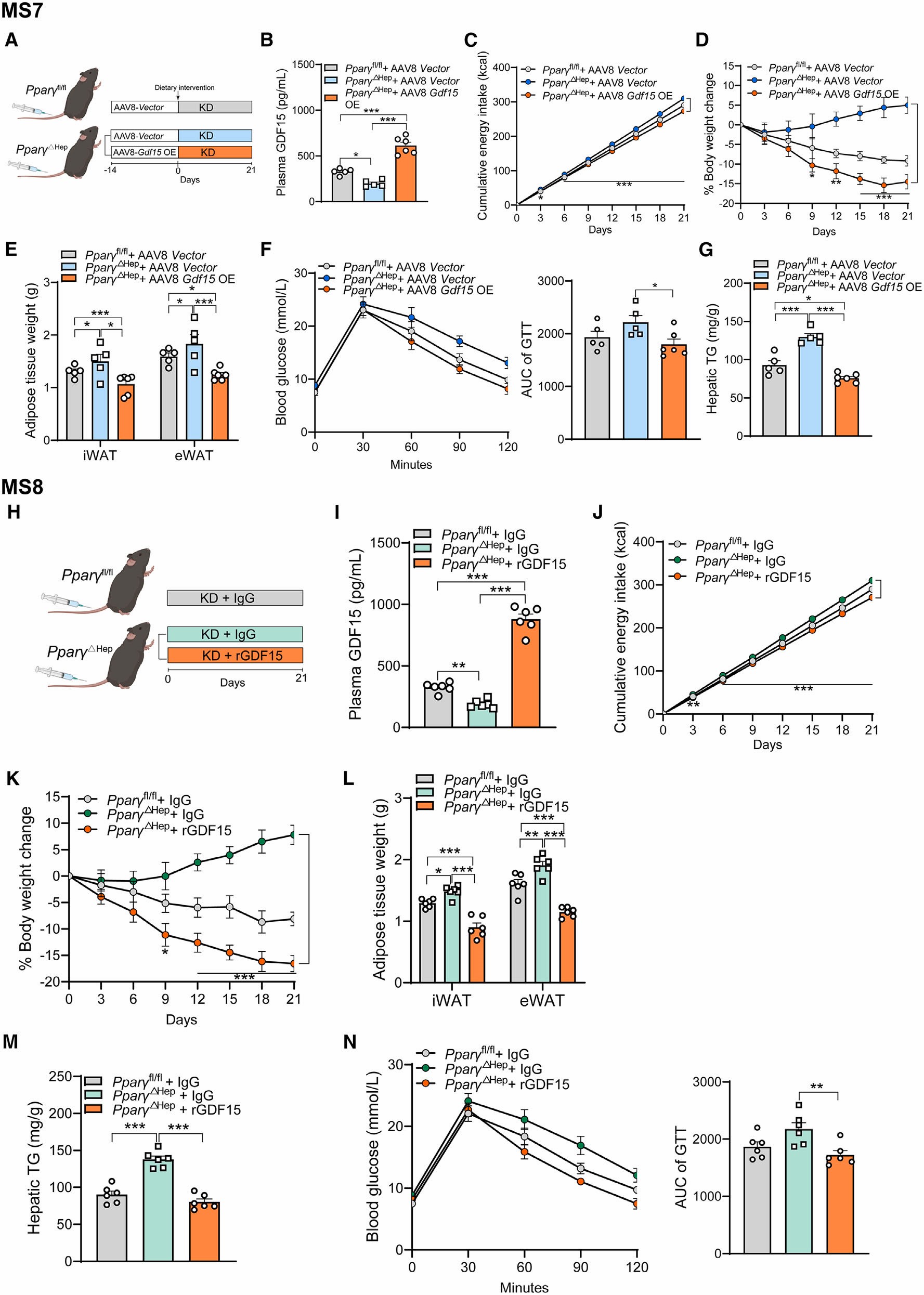
图7 肝脏GDF15过表达或重组GDF15可恢复KD饮食对PPARγ△Hep小鼠肥胖管理的作用
综上所述,本文揭示了KD饮食介导的肥胖管理的作用机制:KD饮食通过激活肝脏组织PPARγ的活性进而促进肝脏GDF15表达和分泌,升高血浆中GDF15水平,最终发挥作用,增加了对KD饮食作用机制的理解,为肥胖的治疗提供了新的见解,也为肥胖管理策略开辟了新的研究方向。
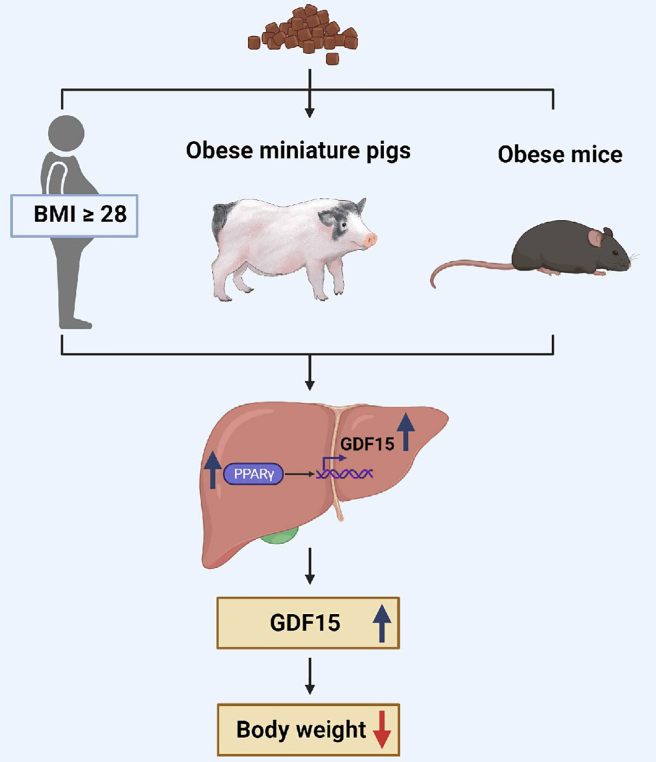
图8 KD饮食介导肥胖管理的分子机制示意图









 1068
1068










